原噬菌体是温和噬菌体感染细菌后,将其自身基因组整合到细菌染色体中的特殊存在形式。它在细菌群落的结构与功能调控中扮演着重要角色。对原噬菌体进行准确鉴定,成为深入研究其功能的首要步骤。然而,这一鉴定工作面临两大挑战:首先,噬菌体基因组具有极高的变异性,导致基于序列相似性的蛋白鉴定方法效力大幅降低;其次,原噬菌体与宿主基因组的边界往往模糊不清,使得整合位点附近的基因难以准确判定是源自细菌本身,还是来源于原噬菌体。
2025年8月20日,生命中心研究员,清华大学基础医学院梁冠翔课题组在《Genome Biology》杂志上发表了一项题为“Highly accurate prophage island detection with PIDE”的研究,该研究开发了一款名为PIDE(Prophage Island Detection using ESM-2)的工具。该工具基于蛋白质大语言模型,能够高效且精准地从宏基因组或细菌基因组中识别原噬菌体区域,同时可以用于病毒组数据中噬菌体基因组的挖掘。为宏基因组与病毒组数据分析提供了基于人工智能模型的有力工具。
该工具的核心思想是,通过使用带有细菌和噬菌体蛋白标签的数据对ESM2模型进行微调实现对细菌基因组中噬菌体蛋白的准确预测,进而采用基于基因密度的聚类算法,精确界定原噬菌体的基因组蛋白表达基因边界(图1)。
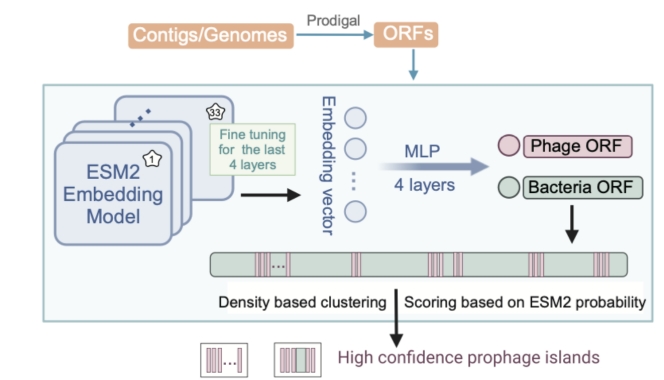
图1 PIDE 流程框架
在与VirSorter2、PHASTER和geNomad等现有高性能原噬菌体/噬菌体识别工具进行比较时,PIDE所识别出的原噬菌体区域不仅与已有工具的结果存在部分重叠,还补充了其他工具未覆盖的区域,展现出PIDE在特异性方面的显著优势,进一步凸显了其与现有工具不同的检测能力(图2)。
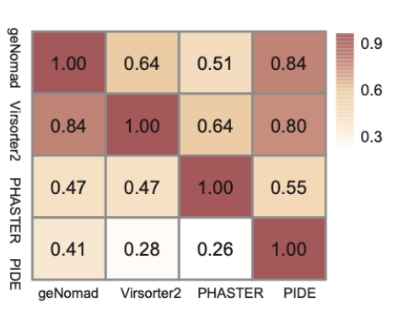
图2不同工具之间预测出的原噬菌体的重合程度
此外,该研究并未依赖传统使用的模拟数据评估工具性能,而是通过细菌菌株原噬菌体诱导病毒组测序,构建了一套准确反映活跃原噬菌体区域的标准实验数据集,为不同工具的评估提供了可靠基准。结果显示,PIDE在原噬菌体边界的碱基水平预测中,在召回率与精确度之间取得了更优的平衡(图3)。
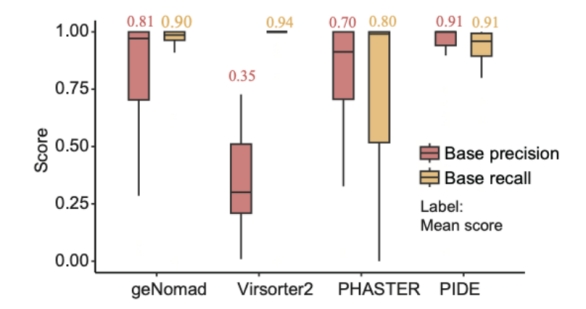
图 3 各工具在碱基水平上对原噬菌体预测精确度与召回率
研究团队进一步将PIDE应用于UHGG 2.0数据库中4744个代表性肠道细菌基因组,发现88.5%的细菌处于溶原状态。通过对原噬菌体编码基因的功能注释,研究揭示这些原噬菌体区域携带的基因可能广泛参与细菌的多个关键生物学过程,包括生长调控、抗生素抗性、代谢途径及毒力等。此外,PIDE基于Uniprot数据库进行微调,具备广泛的适用性,不仅适用于人体肠道微生物组,还可扩展至土壤、海洋等多种生态环境,将为原噬菌体及病毒组的相关研究提供强有力的技术支持。
生命中心研究员、清华大学基础医学院梁冠翔助理教授为本文通讯作者,清华大学基础医学院2020级已毕业博士生高鸿雁、2022级博士生李博文、2021级博士生郭子菡合作完成框架构建、算法整合以及数据产出等工作,为该论文的共同第一作者。清华大学基础医学院2021级博士生郑磊、2023级博士生陈俊楠等也为本研究做出了重要贡献。研究得到国家自然科学基金、清华-北大生命科学联合中心、清华大学启动基金、清华大学笃实基金以及山西医科大学-清华大学医学院前沿医学协同创新基金等支持。
工具主页:https://github.com/chyghy/PIDE
原文链接:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03733-0


