

生命中心PI张强锋课题组利用深度学习人工智能算法分析单细胞ATAC-seq数据
2019年10月8日,生命中心PI张强锋课题组在《自然通讯》(Nature Communications)上发表题为“SCALE方法基于隐特征提取进行单细胞ATAC-seq数据分析”(SCALE method for single-cell ATAC-seq analysis via latent feature extraction)的学术文章。
真核生物的染色质具有复杂的高级结构,由DNA一圈一圈缠绕在组蛋白上形成串珠式模型并进一步折叠聚集而成。基因的转录必须要将相应的染色质打开形成开放区域才能结合其他的转录调控因子。因此可以说染色质开发区域是基因组编码生命的窗口。单细胞ATAC-seq(Assay for Transposase-Accessible Chromatin using sequencing)技术在单细胞层次上通过Tn5 DNA转座酶在开放染色质插入测序接头进行标记并测序,从而获取“高分辨“的单细胞精度的染色质开放图谱,并依此揭示细胞异质性的调控机制。
越来越多的研究者们应用单细胞ATAC-seq技术,在肿瘤、免疫、发育领域获取大量的测序数据。然而,目前没有一个有效的方法可以很好的分析挖掘海量的单细胞ATAC-seq数据中宝贵的生物信息。单细胞ATAC-seq数据分析的难点在于数据本身。第一,细胞整体的染色质开放位点数有几十万之多,造成所谓的“维度灾难”。另外,由于生物的原因许多潜在的开放没有信号,数据异常稀疏,技术限制带来的数据丢失极大程度上加剧了这种现象。特别的,在二倍体基因组上一个开放区域一般至多只有两个拷贝,使得数据近乎二值化。这些问题都给单细胞ATAC-seq数据的分析带来了巨大挑战。
近日,张强锋课题组发表的文章提出了SCALE,利用人工智能深度学习的方法,结合变分自编码器和高斯混合模型,提取单细胞ATAC-seq数据的隐层特征,将问题从复杂稀疏的高维度的染色质开放图谱空间投射到了简单抽象的低纬度特征空间。这种处理不但可以发现和解析细胞特异性的染色质图谱模式,还通过相似细胞信息共享,填补了技术限制导致的缺失值,从而巧妙地解决了单细胞ATAC-seq数据中高维度、稀疏性、二值化等问题。SCALE提供了完整的可视化、聚类、数据增强、帮助下游生物信息的挖掘,为研究者们解码单细胞表观遗传学提供了有力的工具。
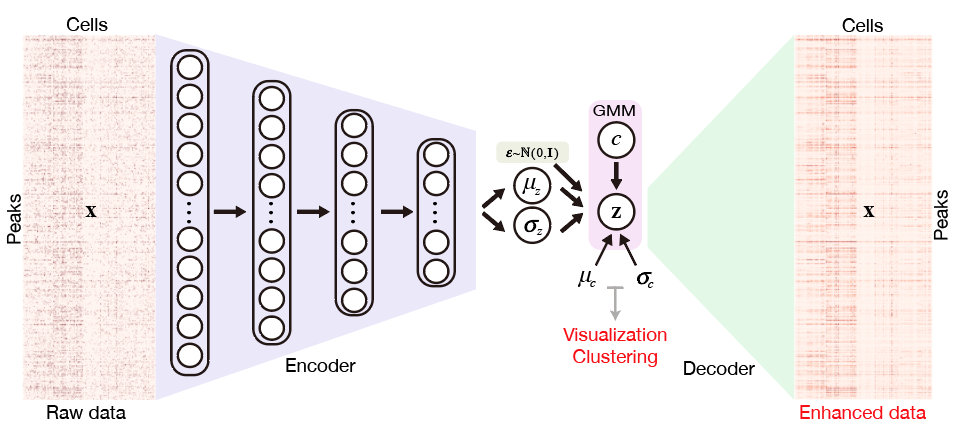
图1. SCALE的模型框架
清华大学生命学院研究员张强锋为本文通讯作者,CLS项目博士生熊磊为本文第一作者。本工作得到了北京大学高歌教授、清华大学张奇伟教授以及加州大学河边分校和清华大学姜涛教授的帮助,并获得国家重点研发计划项目、国家自然科学基金、清华大学结构生物学高精尖中心、清华-北大生命科学联合中心的资金支持。
原文链接:https://www.nature.com/articles/s41467-019-12630-7
